To evaluate the strengths and limitations of GEMDOCK and to compare with several widely used methods (DOCK, GOLD, and FlexX), we first tested our program on docking 10 antagonists back into complexes with experimentally x-ray structures from the Protein Data Bank (PDB).
Second, we tested GEMDOCK on estrogen receptor alpha (ERα), proposed by Bissantz et al. , to evaluate the screening utility of our program.
The docking accuracy of GEMDOCK was comparable with the best available methods and the screening performance was better than that of competing methods.
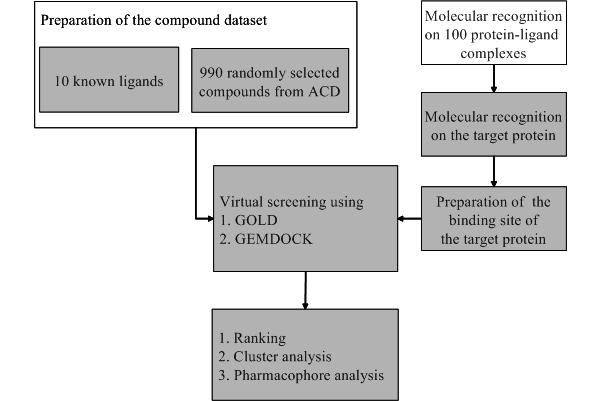
Figure 1. 10 known ERα antagonists
Figure 2 shows the binding-site pharmacophores and ligand preferences that were identified by superimposing 4 crystal structures of ERα shown in Figure 1 (1err, 3ert, 1hj1 and 1uom).
Three binding-site pharmacological preferences and interactions were identified and circled as A, B, and C. The dash lines indicate the hydrogen bonds formed between the ligand and binding site.
According to these observations and docking results, we added following pharmacological weights: D351-OD1, D351-OD2, E353-OE2 and R394-NH2 are hydrogen bonds with weighted value 3.0; H524-ND1 are hydrogen bonds with weighted value 2.0.
These weights were used for calculating the value Epharma for ER ligand preferences, the number of electrostatic atoms was set to 2 because all antagonists (Figure 1) have no charged atoms.
If the number of charged atoms inside a compound is more than 2, WPelec will be 20.
The ligand hydrophilic preference (WPhb) was assigned according to the ratio as
rhb = (NHbond) / (Ntotal - NHbond)
where NHbond is the number of polar atoms (i.e., the atom type is donor, acceptor, or both). Ntotal is the total atom number of the ligand. If rhb is more than 0.35, WPhb was set to 20.
Figure 2.
Binding-site pharmacophores identified by superimposing four crystal structures of ERα.
GEMDOCK executed 3 independent runs for each complex. The solution with lowest scoring function was then compared with the observed ligand crystal structure. We docked each ligand of four antagonists (Figure 1) back into its native complex and the reference protein (3ert). We based the results on root mean square deviation (RMSD) error in ligand heavy atoms between the docked conformation and the crystal structure. The results were shown in Table 1. During flexible docking GEMDOCK obtained similar results whether the pharmacophore preferences (i.e. Epharma and Eligpre) were considered or not. The RMSD values of docked conformations were less than 1.5 angstroms when docking into their native binding site. On the other hand, when docking into the binding site of the complex 3ert.OHT, two ligands could not be successfully docked into the x-ray poses because the side-chain conformation of HIS524 in the reference protein 3ert differs from other complexes. The performance of GEMDOCK was better than GOLD on this test set except the ligand, 1hj1.AOE. The ligand was well docked in the apolar binding site, with the exception of the long acyclic side chain result in the docking deviation mostly.
|
Table 1. Comprison GEMDOCK with GOLD on docking four antagonists back into its native complex and into the reference bidning site (3ert)
|
| Antagonist name a |
No. of polar atoms b |
No. of hydrogen bonds c |
GEMDOCK |
GOLD |
| Binding site: native |
Binding site: 3ert |
Binding site: native |
Binding site: 3ert |
| pharma. weight (yes) |
pharma. weight (no) |
pharma. weight (yes) |
pharma. weight (no) |
Raloxifene
(1err.RAL)
|
5 |
4 |
0.66 |
0.65 |
1.37 |
2.36 |
1.02 |
1.68 |
Tamoxifene
(3ert.OHT)
|
3 |
2 |
0.60 |
0.75 |
0.60 |
0.75 |
1.15 |
1.15 |
ICI-164384
(1hj1.AOE)
|
4 |
3 |
1.41 |
1.05 |
4.15 |
5.35 |
5.07 |
3.92 |
Tetrahydroisochiolin
(1uom.PTI)
|
4 |
4 |
0.80 |
0.43 |
0.89 |
0.85 |
0.56 |
1.56 |
a the four characters and three characters separated by a period denote the PDB code and the ligand name in PDB respectively.
b the number of the atoms that may form a hydrogen bond; i.e., the atom type is one among the three types, both, donor, and acceptor.
c the number of hydrogen bonds formed between the ligand and protein was derived from the native crystal conformations based on our scoring function.
|
Figure 3 shows the results of GEMDOCK using different combinations of pharmacophore preferences that are ligand preferences (Eligpre) and binding-site pharmacophore (Epharma). We tested GEMDOCK on different combinations to evaluate the performance and search behavior. GEMDOCK generally improves the screening quality by considering both ligand preferences and binding-site pharmacophore weights although we did not attempt to refine any parameters of these combinations.
Figure 3.
GEMDOCK results for true hits against ranks.
Table 2 compares GEMDOCK with four docking methods (Surflex, DOCK, FlexX, GOLD) on the same target protein and screening database at true positive rates ranging from 80% to 100%. For GEMDOCK on the target ERα, the ranks of the 10 antagonists were 1-8, 13 and 19. For the true positive rate of 100%, the FP rate for GEMDOCK is 0.9%. In contrast, the FP rates for competing methods are 2.9% (Surflex), 18.9% (DOCK), and 23.4% (GOLD). FlexX couldn't find the docked solution of RU-58668 (EST09) and the FP rate is higher than other approaches. Besides the FP rate of GEMDOCK is the lowest among these five approaches.
|
Talbe 2. Comprison of GEMDOCK with four methods by false positive rates on screening 1000 compounds
|
| True positive % |
GEMDOCK |
Surflex |
DOCK |
FlexX |
GOLD |
80 |
0.0 a (0/990) |
1.3 |
13.3 |
57.8 |
5.3 |
90 |
0.4 (4/990) |
1.6 |
17.4 |
70.9 |
8.3 |
100 |
0.9 (9/990) |
2.9 |
18.9 |
- b |
23.4 |
a the false positive rate from 990 random ligand (%).
b FlexX can not yield the docked solution for RU-58668 (EST09)
|
|
